


Les céroïdes lipofuscinoses neuronales (CLN) sont des maladies neurodégénératives sévères rattachées aux maladies lysosomales. Un certain nombre de gènes responsables de ces affections ont été, et continuent à être, découverts (nommés CLN1, 2, 3 etc…). Ces maladies se caractérisent cliniquement par une épilepsie myoclonique (avec secousses musculaires), un retard psychomoteur et une perte progressive de la vision. Elles existent sous quatre formes cliniques principales, définit selon l’âge habituel de début des signes : infantile (CLN1), infantile tardive (CLN2, CLN5, CLN6, CLN7, CLN8), juvénile (CLN3, CLN12) et adulte (CLN4, CLN11, CLN13) avec l’existence d’importants variants (par exemple, occasionnellement certaines mutations dans le gène CLN1 amènent à une évolution de la maladie plus proche de celle observée chez des patients CLN3).
Différentes stratégies thérapeutiques sont explorées en vue d’offrir une option de traitement pour l’évolution inexorable des symptômes de ces maladies.
En 2017, le premier traitement spécifique pour des patients CLN2 obtenait une autorisation de commercialisation des agences américaine et européenne du médicament. Développée par le laboratoire BioMarin, la cerliponase alfa (nom commercial Brineura™) est une enzyme recombinante. Elle est injectée toutes les 2 semaines directement au niveau du cerveau des malades afin de venir suppléer l’enzyme naturelle défaillante. Les résultats publiés des études cliniques montrent que le traitement permet un ralentissement spectaculaire de la perte de la marche et du langage chez les enfants traités. Ce traitement est proposé aux malades ayant conservés des facultés motrices et verbales.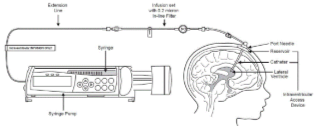
Des essais cliniques basés sur une stratégie de thérapie génique, sont aujourd’hui en cours.
Pour la forme infantile tardive impliquant le gène CLN2 (la plus commune en France), un premier essai clinique a été mené aux Etats-Unis, basé sur l’injection, directement dans le cerveau, d’un vecteur de type AAV2 contenant le gène CLN2. Le traitement, administré en une fois, a permis d’observer sur les imageries du cerveaux un léger ralentissement de la progression des atteintes. L’effet étant modéré, un second essai de phase I/II utilisant un vecteur de type AAV10 et ayant pour objectifs d’évaluer la sécurité, une éventuelle toxicité et l’efficacité du traitement a débuté en 2010. L’espoir est que ce nouveau vecteur permettra d’améliorer les résultats thérapeutiques. Un troisième essai reprenant le même protocole mais élargissant les critères d’inclusion des patients est également en cours.
Pour la forme infantile tardive impliquant le gène CLN6, une étude de l’histoire naturelle de la maladie faite chez 14 malades a montré que sur une période de 24 mois, les malades perdent de 2 à 3 points au test simplifié de Hambourg. Ce test permet de coter de 0 à 3 la fonction motrice et la fonction verbale (0 = plus de fonction, 3 = fonction normale), donnant un score maximal de 6 lorsque les deux fonctions sont normales. En 2016, un essai clinique de phase I/II, utilisant un vecteur de type AAV9 pour apporter des copies non défectueuses du gène CLN6 aux cellules est débuté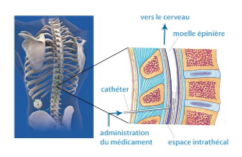 . Le vecteur médicament est injecté en un traitement unique par voie intrathécale (injection au niveau des lombaires, dans l’espace contenant le liquide céphalo-rachidien qui baigne le cerveau et la moelle épinière). En septembre 2018, le laboratoire Amicus acquière les droits exclusifs sur la poursuite du développement de cette thérapie.
. Le vecteur médicament est injecté en un traitement unique par voie intrathécale (injection au niveau des lombaires, dans l’espace contenant le liquide céphalo-rachidien qui baigne le cerveau et la moelle épinière). En septembre 2018, le laboratoire Amicus acquière les droits exclusifs sur la poursuite du développement de cette thérapie.
Début août 2019, le laboratoire publie un communiqué de presse annonçant « des résultats intermédiaires positifs pour la thérapie génique par AAV chez des enfants ayant une céroïde lipofuscinose neuronale de type 6 ». Le communiqué indique que le traitement est généralement bien toléré chez les 12 patients inclus dans l’essai (suivi post traitement entre 6 et 39 mois). Les 8 premiers enfants traités ont un temps de suivi suffisant (16 à 25 mois post-traitement) pour permettre une comparaison de l’évolution de leur score au test simplifié de Hambourg, à celle obtenue dans l’étude de l’histoire naturelle. Chez 7 enfants, le traitement a induit la stabilisation ou un changement initial de +1 ou -1 suivi d’une stabilisation du score. Le patient qui n’a pas été stabilisé dans ses pertes était le plus âgé au moment du traitement (79 mois).
Dans la forme juvénile liée au gène CLN3, plus commune chez les anglo-saxons, là encore le laboratoire Amicus a acquis les droits sur le développement d’une thérapie génique par AAV9 dont l’essai clinique de phase I/II a été lancé aux USA en décembre dernier. L’injection du vecteur médicament se fait par voie intrathécale en une fois. Prévu pour inclure 7 patients, un premier groupe de patients reçoit le vecteur médicament à une dose dite basse, le second groupe à une dose dite haute après évaluation de la tolérance à la dose basse chez les patients du premier groupe. Les objectifs sont d’évaluer la sécurité, l’éventuelle toxicité et l’efficacité de ce traitement. Le recrutement des patients est toujours actifs.
A l’étape préclinique, de nombreuses recherches thérapeutiques sont à l’étude pour différentes formes de CLN malgré le fait qu’on connaisse encore mal le rôle de la plupart des protéines « CLN ».
Le laboratoire Amicus indique poursuivre des travaux précliniques d’un traitement par thérapie génique pour la CLN8 et s’intéresser à initier des travaux similaires pour la CLN1.
Un autre laboratoire, Abeona qui mène actuellement des essais cliniques de thérapie génique pour les maladies de Sanfilippo A et B, porte un intérêt pour les CLN1 et CLN3.
D’autres voies d’approches se concentrent sur la thérapie par des « petites molécules ». Ces substances biologiques ou pharmaceutiques à prise orale peuvent agir à différents niveaux du fonctionnement cellulaire.
Un essai clinique avec une combinaison d’utilisation de bitartrate de cystéamine et de N acetylcysteine a été mené chez des patients CLN1. Des études chez des souris modèles (souffrant d’une CLN) avaient permis d’observer une réduction de la surcharge. Neuf patients ont été traités. Malgré l’observation d’une réduction de la surcharge, il n’a pas été possible de démontrer un ralentissement de la maladie.
Plusieurs études cherchent à mieux réguler l’inflammation des neurones qui est une caractéristique importante dans ces maladies. Un essai clinique initié en 2011 chez des patients CLN3, pour explorer l’impact d’une molécule (le mycophenolate de mofétil) n’a malheureusement pas été concluant.
L’action de molécules visant à stimuler le processus de « fabrication » de lysosomes et restaurer l’autophagie (forme de nettoyage cellulaire effectué par les lysosomes) est également explorée. Des études précliniques aux résultats encourageants pour la CLN3 ont été en partie financée par l’association américaine Beyond Batten Disease qui espère conclure prochainement un partenariat avec une société de biotechnologie (Theranexus) afin de permettre rapidement la poursuite de l’évaluation de cette thérapie en clinique.
[/Delphine GENEVAZ/]






")





